
国家药监局对新冠口服药的审评标准,终于有所改变:主要终点规定为临床改善,次要终点为病毒学指标。
之前的审评标准,临床终点设计是 “住院或死亡患者比例”。不过,在奥密克戎毒株大流行时代,要做出抑制重症患者发展、改善轻症患者症状的临床结果,相当困难。辉瑞和盐野义都未能成功。
此前,业内认为:一直没有国产新冠口服药获批,与CDE指引中的临床终点设置也有关联。
近日阿兹夫定的获批,与这次审评标准的改变,或意味着更多国产新冠口服药上市的加速。
01、这版审评标准有何主要变化?
8月1日,中国国家药监局审评中心发布了一则《关于新型冠状病毒新流行株感染抗病毒新药非临床和临床评价标准的问与答》(下文简称“问与答”)的通知,核心内容是:
“针对奥密克戎变异株,其致病力有所减弱,发展成为重型/危重型或死亡的比例较低,可考虑选择临床疗效指标(在适当的时间内评估至持续临床恢复的时间)的改善作为主要疗效终点,病毒学指标作为关键次要疗效终点。”
相比于CDE在今年2月份发布的新型冠状病毒临床试验技术指导原则(下文简称《指导原则》),这次的“问与答”里,将“临床疗效指标”列为主要终点,并将“病毒载量”上升到了次要终点级别。
虽然2月份发布的《指导原则》里,并没有说“缓解重症率和死亡率”就是新冠口服药审评审批的“金标准”。
但在实际情况中,多位业内人士在今年4月曾表示,新冠口服药Ⅲ期临床的国家药监局审批标准,依旧是以“改善重症、住院率”和“改善症状”为临床主要终点。如果不按照以上指标做临床终点,基本不会被获批做Ⅲ期临床。
而奥密克戎成为COVID-19的主要毒株后,由于感染人群大多是无症状感染者和轻症,重症和住院人数占比很少,这使得重症受试者的入组极为艰难。这导致很多本土新冠小分子项目临床难以进展,被业界认为是国产口服药长期无进展的重要原因。
02、“临床症状改善”,判断难点在哪里?
CDE于2月初发布的那版《指导原则》,已带有一些前瞻性,提到:“随着随机对照试验等其他信息的出现,预期标准治疗可能会发生变化。”
奥密克戎株流行后,全球范围内绝大多数都是轻型/普通型患者,对于新冠口服药的研究,临床终点都向“至持续临床恢复的时间”这个标准上聚集,也就是临床症状改善。
一大问题是,试验中要判断怎样算“临床症状改善”?
对于“临床症状改善”的定义,《原则》也做了一些解释,比如包括临床症状(呼吸困难、发烧等等)、影像学、病原学在内的一些具体指标。
不过,奥密克戎的临床症状,在不同人身上,可能千差万别。
一位业内人士提到:“这个标准虽然有,但是到了临床,往往人为干扰因素很多,比如‘乏力’、‘胸闷’等等,每个人感受不一样,这里面存在一些主观的判断。”
在“临床症状改善”这一点上,无论是辉瑞的Paxlovid、还是盐野义的S-217622,都没有做出阳性结果。
君实按照《原则》指示的方向,和Paxlovid做了一个针对“临床症状改善”的头对头实验,根据其公布的三期结果,显示其在“和P药等效性上”达到了预期终点。而从阿兹夫定的上市来看,似乎只有这一款药目前在这一点上做出了预期的数据。
接下来,还需要观察后续国产口服药将如何做出这个数据。
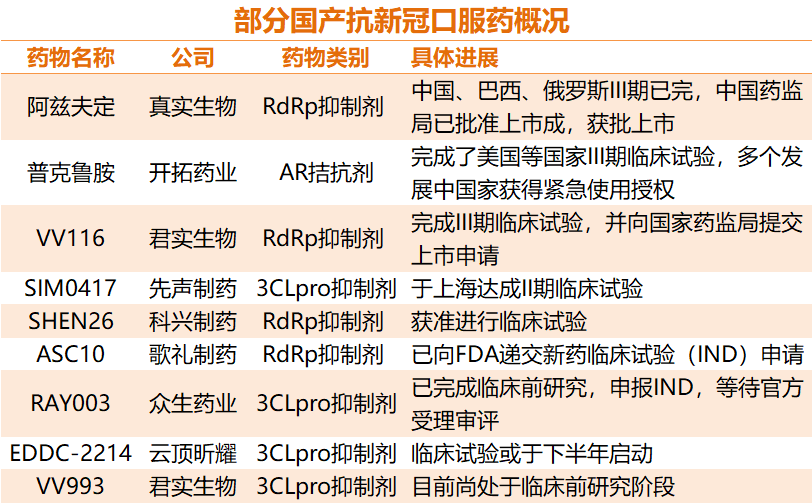
部分国产新冠口服药概况
来源:光大证券
03、病毒载量指标有何重要意义?
这版“问与答”,将“病毒载量”上升到了次要终点级别。关于这项指标,曾有一些争议。
早在今年年初,防控专家吴尊友曾公开提及:“病毒载量不属于三期临床试验的主要指标,三期临床试验指标的选择应当还是取决于对疾病严重情况的观察。临床难做不应该是药监局降低审评标准的理由。”
支持者的观点是“降低病毒载量”和“新冠症状缓解”二者之间没有*的因果关系:“每个人的个体情况不同,有的人病毒载量低,但症状严重;而有的人恰恰相反。”
但曾在GSK、默沙东工作多年的药理及药物安全学家曾皓宇认为,“降低病毒载量”这一指标,对于高传染性、低住院率和重症率的奥密克戎毒株是具备重要意义的。
他认为:“降低病毒载量这一指标,在阻断社区传播上很重要。感染奥密克戎毒株的很多人是无症状感染者或轻症,改善症状的指标意义有限。但降低病毒载量到无法传播的水平,非常适合现阶段中国的防控思路。”
04、病毒在变异,审评标准也在不断更新
药品的审评是在科学和逻辑的基础上,基于某一个“标准”做出程序化的审批,这个过程是人做的,不是机器,这个标准有一个“解释空间”,不是严格的0和1的区别。
新冠病毒在变异,审批标准也在不断更新。
考虑到这两年,大湾区和长三角两个药审的分中心成立,CDE和产业界的交流互动越来越多,关于新冠口服药临床终点的“标准”,药监一直在和相关药企不断增加技术层面的沟通。
而阿兹夫定的获批,也说明了药监其实已经在基于新的新冠口服药审批标准做事。
但这一次药监把新的标准,以官方文件的形式发在了药监局官网上,也是在向整个业界发出信号:从国家药品审批的角度来看,新冠口服药的临床标准确实需要根据疫情形势变化。
2020年12月,在新冠病毒*次变异时,BioNtech的负责人乌古尔·萨欣(Ugur Sahin),曾表示:“可在六周内提供变异后的疫苗。”
新冠病毒变异很快。得益于生物医药产业的发展,当前的应对手段迭代更新也能很快。但是,监管因为其程序的重要性,速度不一定能赶上前者。
不过,监管程序也并不是没有改善的空间。
当然,“标准的变化”不等于“标准的松懈”,如果只是一味放低标准就能解决问题,那也不需要监管了。重要的是,如何在标准降低之下,来找到一套执行程序,去满足新药审批的科学性和逻辑性。
【本文由投资界合作伙伴微信公众号:深蓝观授权发布,本平台仅提供信息存储服务。】如有任何疑问,请联系(editor@zero2ipo.com.cn)投资界处理。





