
10月30日,Sarepta Therapeutics公布全球*治疗杜氏肌营养不良症(DMO)的一次性基因疗法Elevidys三期验证性临床EMBARK(SRP-9001-301研究)的初步数据,该研究没有到达NSAA评分(北极星动态评估,运动功能测量指标)主要临床终点。
今年6月,Elevidys通过FDA批准上市,用于4-5岁、可独立行走的杜氏肌营养不良症(DMD)儿童(在外显子8和/或外显子9上存在缺失突变的DMD儿童中禁用)。随后,这款定价为320万美元(约合人民币 2341 万元)一针的新药迅速跻身上榜,成为了当前全世界第二贵的药。
不过,根据当时的资料来看,Sarepta 一开始的目标是将Elevidys的使用人群定在全年龄段,而FDA加速批准却将其圈定在4-5岁的儿童中。此次实验,一是为了确认Elevidys的临床有益,其次便是尝试与FDA协商以扩大适用人群。据Elevidys随后在公开信内的回应,他们希望率先将年龄扩展至4-7岁,未来在疾病阶段、抗体情况、基因突变类型等方面继续扩展适应症。
到达所有关键次要终点
DMD是常见的X连锁隐性遗传性肌肉变性疾病,在男性新生儿中的发病率约为1/3500。通过基因检测,93.1%的患者具有遗传学病因,为早期的基因治疗奠定基础。由于基因突变,患者肌肉组织缺少抗肌萎缩蛋白,致使肌肉无力并渐进性肌肉损失。大多数患儿在12岁时都无法行走,并会在30岁左右死亡。
Elevidys获批之前,DMD在临床上主要以对症治疗以及支持治疗为主,例如激素和激素代替疗法等。但是这些疗法需要长期给药,只能缓解疾病症状,不能针对疾病根源。
Elevidys是一种基因替代疗法,由罗氏与Sarepta 合作开发。Elevidys通过在肌肉组织中定向产生抗肌萎缩蛋白的功能成分来解决DMD的根本原因;将截短了的抗肌萎缩蛋白(迷你DMD)的转基因包装在AAV病毒载体中,单次静脉注射使得患者肌肉生成具有部分抗肌萎缩蛋白功能的重组蛋白,可以对携带任何类型DMD致病基因变异的患者生效。
今年5月,FDA咨询委员会以8:6的微弱优势赞成Elevidys加速批准上市;6月,FDA批准Elevidys作为DMD的全球*基因疗法,但由于Sarepta将Elevidys表达的微抗肌萎缩蛋白水平作为替代终点,因此FDA要求完成验证性临床研究以证实临床获益。
这项验证性临床试验,是一项针对4至7岁DMD患者的全球随机、双盲、安慰剂对照的3期临床研究。据Sarepta 披露,试验共有125例患者入组,1:1随机分配接受Elevidys或安慰剂治疗,主要终点为NSAA评分,次要终点包括蛋白表达水平、TTR、10MWR等。
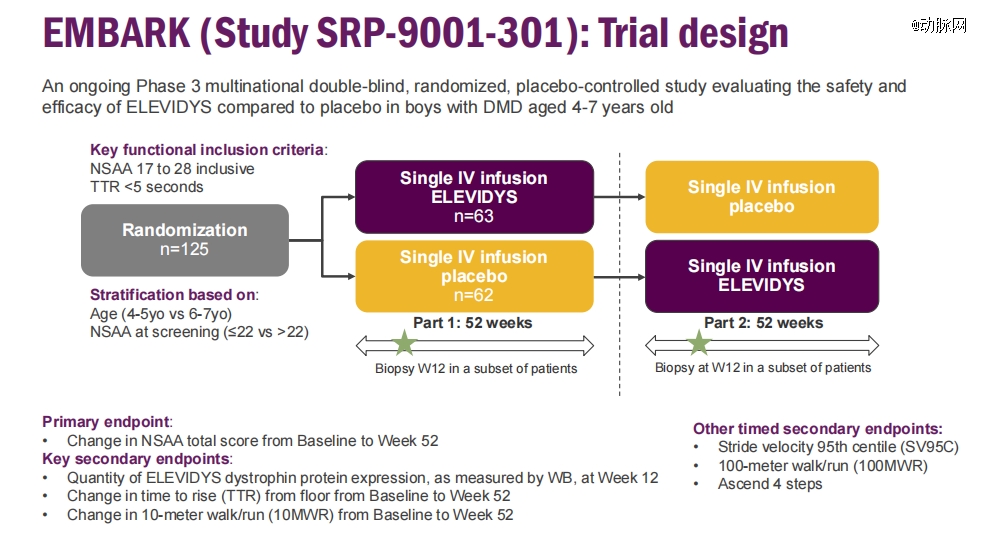
数据显示,在EMBARK中,与接受安慰剂治疗的患者相比,在第52周时,接受Elevidys治疗的参与者的NSAA评分有所提高,但并未到达预定的主要临床终点(趋势改善但没有达到统计学显著差异)。
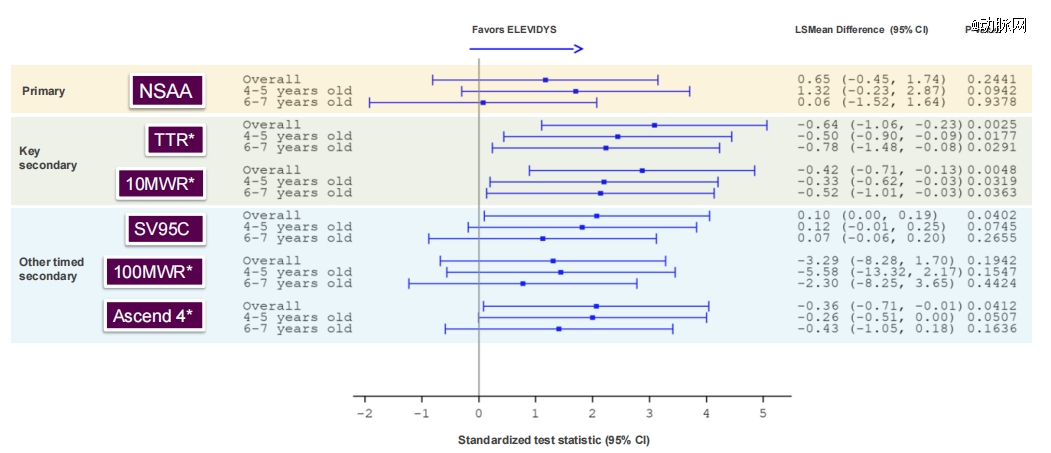
同时,所有关键预先指定的次要终点,包括起身时间(p=0.0025)(TTR)和10米步行/跑步测试(p=0.0048)(10MWR),均获得稳健且具有统计意义的提升,成为其具有临床意义治疗益处的证据,且其在所有年龄段都具有统计意义。
另外,EMBARK试验没有观察到新的安全信号。在125名受试者中,仅有7名受试者经历了与治疗相关的严重不良事件,其严重程度与此前的实验数据一致。

未到达主要终点:撤销还是扩大适应症?
2019年,罗氏以11.5亿美元收购Elevidys在美国以外的*经销权。根据协议,罗氏将支付Sarepta7.5亿美元预付金,并动用4亿美元认购其股份。一旦Elevidys获批并达到一定销售成绩,Sarepta可收取的款项最多可达17亿美元。
这项被罗氏寄予厚望的罕见病新药,此前便已获得FDA快速通道认定、罕见儿科疾病(RPD)认定,获得美国、欧盟、瑞士和日本孤儿药认定(ODD)。
Elevidys的加速批准是基于骨骼肌中微型肌营养不良蛋白表达的增加。据公开信息,Sarepta在2期临床试验到达临床终点后便率先申报,所提交的生物制品许可申请(BLA)包括3项临床研究的疗效和安全性数据:Sarepta所提交的生物制品许可申请(BLA)包括3项临床研究的疗效和安全性数据,以及这3项临床研究的综合分析,比较了功能结果与倾向性评分匹配的外部对照(EC)。
一般来说,如果临床试验未能到达主要重点,FDA不会认为次要研究目标的数据是积极的。也就是说,根据FDA的加速批准路径,药物可以通过单臂临床试验、替代终点获批上市,满足一些紧迫的临床需求,但必须满足两个条件:一是企业要按照与FDA达成的协议,在协定的时间内,完成协定的临床确认性试验;二是若达到预期的临床获益与风险比,FDA会将加速批准提升为彻底批准,否则将“加速”撤销加速批准。
据《英国医学杂志》2021年的一项研究发现,自FDA在1992年建立其加速批准药物途径以来,在253种获批药物中,近一半的药物尚未被确认为临床有效,对FDA数据的进一步分析发现,只有16种通过该途径获得批准的药物被撤回,其中大多数药物被证实缺乏疗效,甚至在某些情况下,这些药物尚未进行过验证性试验。
近年来,由于多款加速批准的创新药在验证性三期临床中失败,FDA加速批准程序正在收缩,随着Robert Califf二度出任FDA局长,包括加速撤回在内的新框架呼之欲出。
不过Sarepta方面对此次EMBARK试验结果持积极观点,据Stat报道,Sarepta已经与FDA高级官员,包括生物评估和研究中心(Center for Biologics Evaluation and Resaerch)主任会面,其CEO Doug Ingram表示,“FDA将迅速审查新数据,并完成对适应症标签扩展的审查。”
如此来看,说药物会被“撤销”恐怕是无稽之谈。Sarepta 随后在其社群公开信中回应关于Elevidys后续扩展适应症方面的问题,包括年龄、疾病阶段、抗体情况、基因突变类型。重点包括:
● 除了目前 FDA 已批准的适应症和 EMBARK 研究覆盖的人群之外,Sarepta还在进行一项全球 3 期临床研究ENVISION (NCT05881408),旨在评估 Elevidys对于可独立行走男孩(任何年龄)和较大年龄卧床男孩(8-18 岁)的安全性和有效性。
● 预计将开始两项不同的研究,以探索两种不同的机制来减少预先存在的 AAVrh74 抗体。
【本文由投资界合作伙伴动脉网授权发布,本平台仅提供信息存储服务。】如有任何疑问,请联系(editor@zero2ipo.com.cn)投资界处理。





